Limiting Evergreening for Name-Brand Prescription Drugs
To encourage medical innovation, the Food and Drug Administration (FDA) grants temporary market exclusivities to new brand name drugs. These exclusivities prohibit generic drug competitors from accessing the market for a limited period. However, drug manufacturers are often able to take advantage of the current rules, using “evergreening” strategies to extend their exclusivity periods and either delay generic drug market entry or limit the number of patients who switch to a new generic.
One evergreening tactic manufacturers employ involves introducing a new “line” or version of their drug shortly before a generic competitor is released. This new line can be granted its own exclusivity period. For example, a manufacturer may introduce an extended-release formulation just before a generic of the original immediate-release formulation enters the market. This can allow a brand manufacturer to maintain market share in the face of generic competition—increasing its profits and increasing payer and patient costs.
As part of the Health Savers Initiative, this paper examines a policy option to prevent evergreening delays of generic drug competition through new FDA exclusivity rules. This could lead to meaningful savings for consumers, commercial insurers, and government payers. The policy change could also speed up the market entry of brand extended-release and other reformulations, providing clinical benefits to patients.
Over the next decade (2021-2030), this policy could:
- Reduce federal deficits by at least $10 billion
- Save Medicare Part D $7 billion in drug costs and Medicare beneficiaries $4 billion in lower premiums and cost sharing
- Reduce federal and state Medicaid drug spending
- Reduce private sector drug costs by $9 billion
Given the high and rising costs of health care, a number of bold policy changes will be needed to assure long-term affordability and sustainability. Limiting the ability of drug manufacturers to delay generic competition should prove an attractive option to policymakers.
The Health Savers Initiative is a project of the Committee for a Responsible Federal Budget, Arnold Ventures, and West Health, which works to identify bold and concrete policy options to make health care more affordable for the federal government, businesses, and households. This brief presents an option meant to be just one of many, but it incorporates specifications and savings estimates so policymakers can weigh costs and benefits, and gain a better understanding of whatever health savings policies they choose to pursue.
The Need for Action
National retail pharmaceutical spending has risen dramatically over the last two decades, from $122 billion in 2000 to $370 billion in 2019.1 While retail drug costs have been a generally consistent share of total health care expenditures—around 10 percent—both drug spending and overall health care spending has significantly outpaced the growth of the US economy.
Moreover, while the out-of-pocket prescription drug cost per patient has generally declined over the last decade in aggregate, a significant number of patients face very high out-of-pocket costs.2 For example, in 2017, one million Medicare Part D beneficiaries were above the catastrophic threshold and had average out-of-pocket costs above $3,200, double the number of seniors who were above the catastrophic threshold a decade earlier.3 According to the National Health Expenditure Projections published by the Centers for Medicare and Medicaid Services (CMS), future expected growth in total prescription drug spending will exceed 5 percent annually and be attributable to both increases in drug prices and the intensity of usage, particularly among patients with private health insurance. In Medicare, drug spending is projected to grow by close to 8 percent.
High drug costs have led many policymakers to seek to lower prescription drug prices, and a broad range of policies have been suggested, some quite expansive in nature and others more modest. One area ripe for consideration is reining in brand manufacturers’ use of FDA exclusivities to unduly delay generic drug entry.
The Hatch-Waxman Act of 1984 facilitated the rise of the modern generic drug market in the United States. It established FDA-granted market exclusivities to provide temporary monopolies for new drugs to encourage drug development. It also created an abbreviated regulatory pathway for generic drug manufacturers in order to generate competition, most notably by establishing a 180-day exclusivity period for the first generic market entrant.4 Broadly speaking, the generic incentives have worked and generic drugs save the US health care system over $300 billion annually.5
However, brand drug manufacturers are increasingly engaging in evergreening strategies, effectively extending the monopoly life of their products. Between 2005 and 2014, 70 percent of the 100 top-selling drugs were granted an exclusivity period (or a new patent) after FDA approval, with half of these drugs receiving multiple.6 Moreover, between 1995 and 2004, the average period of market exclusivity for new drugs with annual sales greater than $250 million increased more than 20 percent, from 10.3 years to 12.5 years.7
The Case for Curtailing Line Extensions
Brand drug manufacturers use numerous evergreening strategies to extend the duration of their monopolies and delay meaningful generic competition. A common evergreening tactic is to strategically time a line extension. For example, when a manufacturer launches a reformulation of a chemical entity already on the market: first introducing an immediate-release (IR) dosage form and later, close to when a generic is expected to enter the market, reformulating the same drug into an extended-release (ER) form.
This tactic takes advantage of the FDA allowing manufacturers to submit a supplemental application for new drug approval.8 Changes to a previously approved brand drug that affect its active ingredient(s), dosage form, strength, or admission method, can receive a three-year “new clinical investigation” exclusivity.
By strategically timing the launch of a line extension, a manufacturer delays the introduction of a clinically improved reformulation in order to prolong market exclusivity. For example, ER formulations are often more desirable both in clinical impact and for improved patient adherence.9 Thus, in this strategy, manufacturers are thwarting generic competition while also keeping potentially beneficial formulations of the brand drug from patients.
There are multiple examples of brand manufacturers launching line extensions to retain brand market share. One example from our data set involves the HIV drug Viramune (see Figure 1). The initial IR formulation entered the market in 1996, however with generic competition approaching, the manufacturer released an ER version in 2011. Thus, despite market entry of a generic IR alternative in 2012, the brand manufacturer was able to maintain significant market share and profit for an additional three years until an ER generic competitor joined with the IR generic in capturing overwhelming market share.
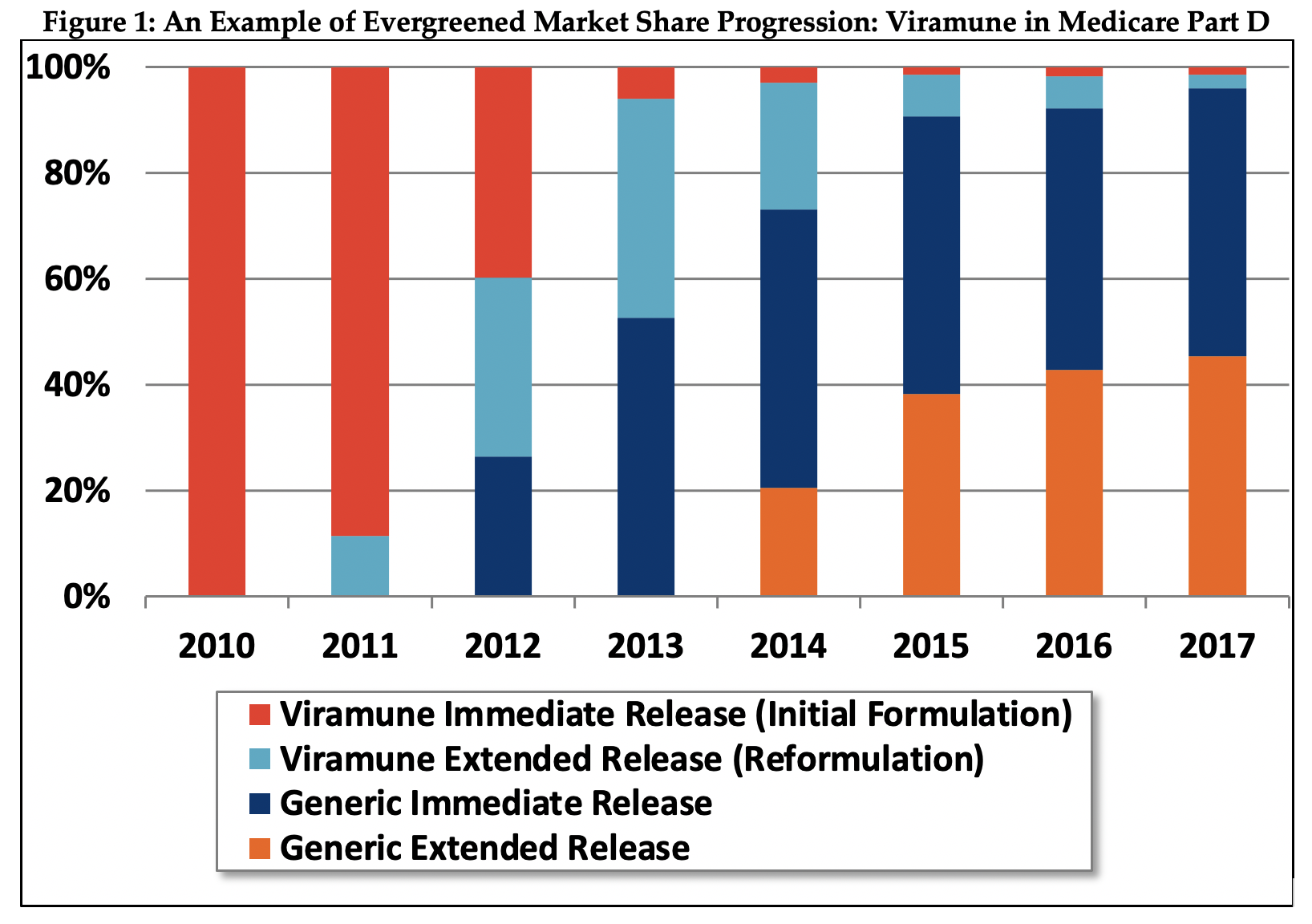
Viramune is a niche HIV drug with a limited usage population overall, however it provides an illuminating example of a full evergreening product progression. Furthermore, the drug’s limited applicability likely made it even more important for the manufacturer to squeeze even a few more years of exclusivity out of the product using the complicated evergreening strategy.
The success of the strategy can be seen even though our data is limited to the Medicare population. We estimate evergreening likely increased Medicare spending by $59 million relative to total Medicare Viramune spending of $149 million over the 2012-2019 period. The drug has higher usage in the non-Medicare population, so the relative increase in commercial market costs of evergreening were likely even higher.
As policymakers and consumers are becoming increasingly concerned about rising prescription drug prices and access to affordable drugs, there have been several legislative proposals introduced on a bipartisan basis, which seek to curtail particular evergreening strategies that take advantage of the FDA drug approval process and/or patent law. In the last Congress (116th), at least five bills of the sort were introduced.10
However, none address the issue of line extensions as an anticompetitive practice like the policy option explored here, which addresses changes to the current framework for FDA-granted regulatory exclusivities.
The Policy Details
Under this policy, manufacturers would be incentivized to introduce extended-release formulations earlier, allowing generic competitors to launch extended-release products sooner, which will promote savings from generic competition in general. The incentives are a combination of limitations to current FDA exclusivities and a new add-on incentive. A more detailed specification of this policy can be found in Appendix A.
This policy option would limit the current three-year FDA market exclusivity that is given for “new clinical investigations” that result in an extended-release or other reformulation. It would prohibit the FDA from approving an application for an evergreened product in the three years prior to expected generic competition (which is normally triggered by patent expiration).
In order to accelerate the availability of clinically improved reformulations, the policy would also provide an extended exclusivity period on the original product if an extended-release or other reformulation is launched within three years of the original product and if the manufacturer demonstrates compelling evidence that the new product would offer substantive clinical benefit.
Products currently on the market would be eligible for an additional three-month exclusivity if an extended-release or other reformulation is launched and/or marketed within three years of the date of enactment of the new policy and, as above, the manufacturer demonstrates compelling evidence that the new product would offer substantive clinical benefit.
This policy would reduce the current incentive to delay the introduction of extended-release and other evergreened reformulations and provide incentives for the early introduction of reformulated products if they can be demonstrated to be beneficial to patients. The add-on exclusivity may also incentivize manufacturers to introduce formulations that provide clinical benefits but are not currently incentivized by the existing regulatory framework.
While the policy is intended to curtail strategic timing of line extensions, it may not be foolproof. Legislation must include clear authority for the FDA to establish rulemaking to define substantive clinical benefit and compelling evidence as well as authority for the FDA to limit the exclusivity extension to only credible applications.
Estimated Fiscal and Financial Impact
Over a decade, we estimate this policy could reduce federal government Medicare Part D costs by roughly $7 billion and beneficiary part D premiums and cost sharing by $4 billion.11 In the initial decade, savings would be derived largely by earlier launches of extended-release products for existing immediate release products and may be less than the long-run savings estimate. In subsequent periods, savings will arise from extended-release products launching concurrently or within three years of new immediate release products, when clinically appropriate. A detailed outline of the methodology and assumptions we used to generate these estimates can be found in Appendix B at the end of this paper.
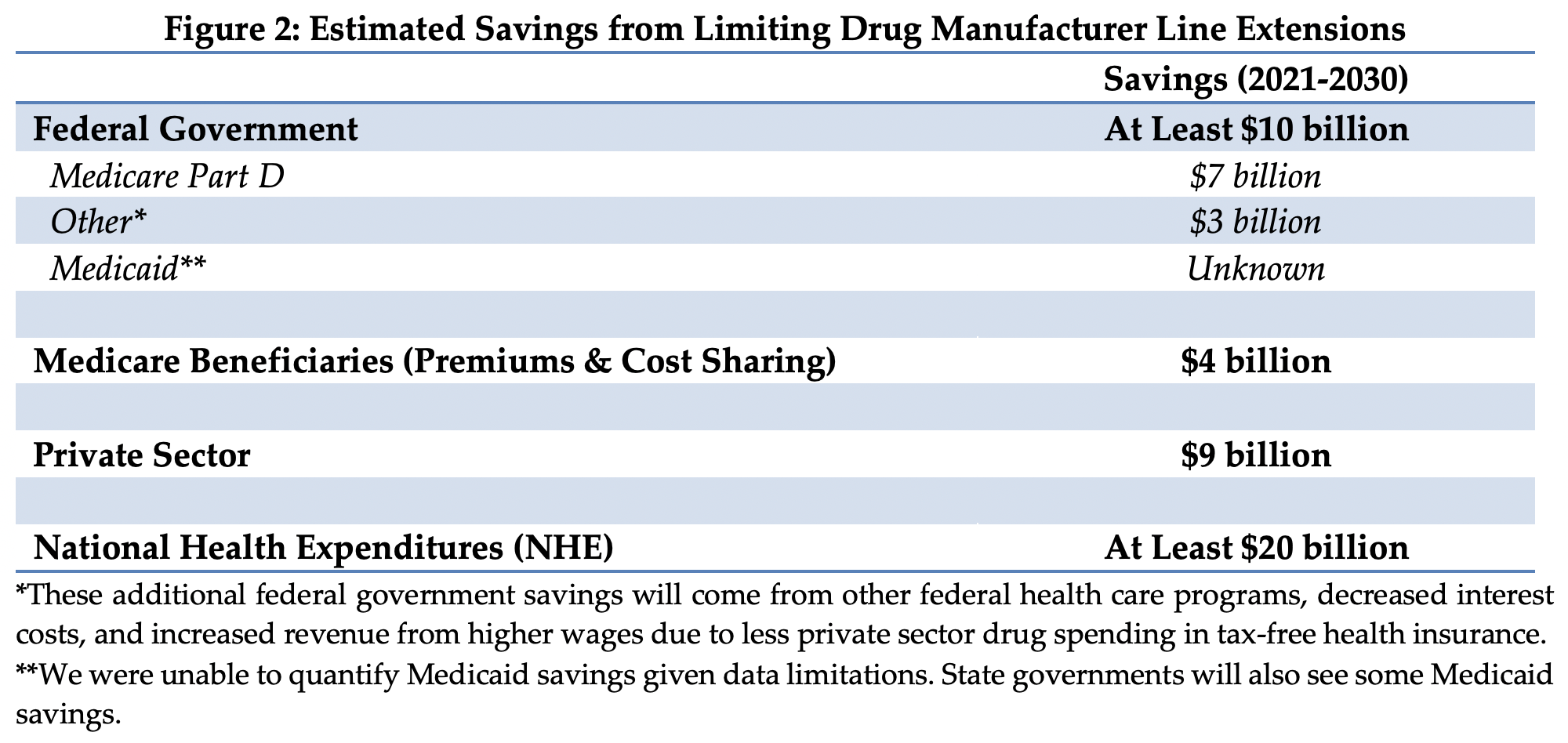
Medicaid would likely see savings under this policy.12 While the Medicaid program realizes substantial rebates on brand drugs, faster generic drug entry would likely represent additional savings. However, due to lack of data on net Medicaid prices for the individual drugs considered in this analysis, we are unable to quantify savings.
There would also be savings in the commercial prescription drug market. We estimate total savings (including out-of-pocket spending) of $9 billion, but this likely understates savings to some degree because our data was limited to total commercial spending already net of rebates to Pharmacy Benefit Managers. These savings would slow the growth of insurance premiums, increase wage growth, and thus generate roughly $2 billion of revenue from higher taxable income.
Appendix A: Policy Specifications
The statutory changes to the Federal Food, Drug, and Cosmetic Act would do the following:
- Define common evergreened product types and prohibit the FDA from approving a supplemental application for an evergreened product in the two years prior to the expiration of the patent that received a Hatch-Waxman extension.
- Provide an extended exclusivity period on the patent of the original product that received a Hatch-Waxman extension if an extended-release or other reformulation is launched within three years of the original product and if the manufacturer demonstrates compelling evidence that the new product would offer substantive clinical benefit.
- Provide an additional six-month exclusivity if the manufacturer of a new drug petitions the FDA before the commencement of Phase 3 trials to produce an extended-release product of a current investigational drug and markets the extended-release product within six months of FDA approval of the original product.
- Provide an additional three-month exclusivity if the manufacturer of an existing drug petitions the FDA within two years after approval to produce an evergreened product of the existing drug and markets the extended-release formulation within three years after FDA approval of the original product.
- Repeal the three-year exclusivity period for reformulations unless that reformulation is introduced in a timely manner. For new drugs, that would be within three years of the approval of the original molecule. For drugs already on the market, that would be within three years of the date of enactment.
Appendix B: Estimating Methodology
The estimates in this document were produced through the joint effort of the partner organizations: The Committee for a Responsible Federal Budget, Arnold Ventures, and West Health.
The starting points for savings estimates were the March 2020 Congressional Budget Office baseline and the March 2020 National Health Expenditure projections from the actuaries at the Centers for Medicare and Medicaid Services. The Heath Savers Initiative will use this pre-Covid-19 baseline for all options to allow for the best comparisons since the full impact of Covid-19 on the baselines is unknown at this time.
For drug savings, we began with Medicare Part D Drug Spending Dashboard data published by CMS,13 which contain spending, claims, and beneficiary counts from 2011 through 2019, and the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations (commonly known as the Orange Book).14
We began by identifying new drug applications (NDAs) along with a dosage form that indicated extended-release, delayed-release, or oral disintegrating. The active ingredient(s) associated with each NDA were used to identify evergreened formulations from the same manufacturer. Corresponding generic versions of both the initial formulation and the evergreened formulation were also identified (if available).
Once the appropriate applications for a given ingredient were identified, we categorized the applications by one of four types: 1) brand initial release (IR), 2) generic initial release (IR), 3) brand extended-release (ER), or 4) generic extended-release (ER). Thirty-seven active ingredients of interest were identified over the nine-year period, and each was matched to a corresponding record from the Medicare Part D Drug Spending Dashboards. Sixteen ingredients contained sufficient data to model savings.
We assume that the new policy proposed here would result in an earlier launch of the ER brand product, and therefore an earlier launch of the generic ER product, thereby yielding savings. We also recognize that there will be some windfall benefit to some manufacturers who would have launched an extended-release product concurrently or within three years of the immediate release drug. We further assume claims for the brand initial release and generic initial release remain constant over time.
We then adjust the savings to account for the additional exclusivity that would be awarded to these products as a reward for the prompt launch of their ER product. Specifically, a portion of the newly switched generic extended-release claims (proportional to the portion of the year that the exclusivity period represents) is switched back to brand extended-release, removing some of the savings for that year. Finally, we transform this analysis from a retrospective review of historical data (2011-2019) to a prospective forecast based on projected trends from the 2020 Medicare Trustees Report.
1 U.S. Centers for Medicare and Medicaid Services, “National Health Expenditure Projections 2019-2028,” May 2021, https://www.cms.gov/files/document/nhe-projections-2019-2028-forecast-summary.pdf.
2 William Carroll et al., “Out-of-Pocket Spending for Retail Prescribed Drugs by Age and Type of Prescription Drug Coverage, 2009 to 2018,” Agency for Healthcare Research and Quality (AHRQ), Statistical Brief #532, December 2020, https://meps.ahrq.gov/data_files/publications/st532/stat532.shtml.
3 Juliette Cubanski et al., “How Many Medicare Part D Enrollees Had High Out-of-Pocket Drug Costs in 2017?” Kaiser Family Foundation, June 2019, https://www.kff.org/report-section/how-many-medicare-part-d-enrollees-had-high-out-of-pocket-drug-costs-in-2017-methods.
4 Hatch-Waxman also added patent term extensions for new drugs to encourage drug development.
5 Association for Accessible Medicines, “Securing Our Access and Savings: 2020 Generic Drug and Biosimilars Access & Savings in the U.S. Report,” 2020, https://accessiblemeds.org/2020-Access-Savings-Report.
6 This is from a comprehensive study by Robin Feldman of patents and exclusivity periods listed in the Orange Book over the 2005–2015 period. This was based on 100 top-selling drugs. Robin Feldman, “May Your Drug Price be Evergreen,” Journal of Law and the Biosciences 5(3), December 2018, pages 590–647, https://academic.oup.com/jlb/article/5/3/590/5232981.
7 Henry Grabowski et al., “Updated Trends in US Brand-Name and Generic Drug Competition,” Journal of Medical Economics 19(9), September 2016, pages 836–844, doi: 10.1080/13696998.2016.1176578.
8 For a rundown of this process see: FDA, “Small Business Assistance: Frequently Asked Questions for New Drug Product Exclusivity,” February 2016, https://www.fda.gov/drugs/cder-small-business-industry-assistance-sbia/small-business-assistance-frequently-asked-questions-new-drug-product-exclusivity.
9 Kimberly Scarsi and Sudan Swindells, “The Promise of Improved Adherence With Long-Acting Antiretroviral Therapy: What Are the Data?,” Journal of the International Association of Providers of AIDS Care (JIAPAC), January 2021, DOI.org/10.1177/23259582211009011; Michael Stirratt et al., “Advancing the Science and Practice of Medication Adherence,” Journal of General Internal Medicine, 33, 2018, pages 216–222, https://doi.org/10.1007/s11606-017-4198-4.
10 The Affordable Prescriptions for Patients Act proposed, among other policy changes, to amend the Federal Trade Commission Act to make it a prima facie antitrust violation for drug manufacturers to discontinue or withdraw a brand drug once it receives notice of an application to market a generic version. The No Combination Drug Patents Act sought to change the non-obviousness standard of patent law with respect to dosing regimens or delivery mechanisms. The Terminating the Extension of Rights Misappropriated Act and the Reforming Evergreening and Manipulation that Extends Drug Years Act both intended to curtail the practice of using later-filed patents to extend product exclusivities. The Reforming Evergreening and Manipulation that Extends Drug Years Act aimed to limit the practice of evergreening and bring generic drugs to market more quickly.
11 Drug spending metrics for Part D drugs are based on the gross drug cost, which represents total spending for the prescription claim, including Medicare, plan, and beneficiary payments. The Part D spending metrics do not reflect any manufacturers’ rebates or other price concessions as CMS is prohibited from publicly disclosing such information. Total dashboard gross costs, which represent spending for the prescription claim, including Medicare, plan, and beneficiary payments, are calculated by using the change in Part D incurred costs and are presented as an approximation.
12 Sean Dickson, “Effect of Evergreened Reformulations on Medicaid Expenditures and Patient Access from 2008 to 2016,” Journal of Managed Care & Specialty Pharmacy, 25(7), July 2019, pages 780–792, doi10.18553/jmcp.2019.18366.
13 U.S. Centers for Medicare and Medicaid Services, “Medicare Part D Drug Spending Dashboard and Data,” February 2021, https://www.cms.gov/Research-Statistics-Data-and-Systems/Statistics-Trends-and-Reports/Information-on-Prescription-Drugs/MedicarePartD.
14 U.S. Food and Drug Administration, “Orange Book Data Files,” February 2021, https://www.fda.gov/drugs/drug-approvals-and-databases/orange-book-data-files.